Catalyst optimization techniques are often based on the qualitative and inductive predictions of chemists screening data.
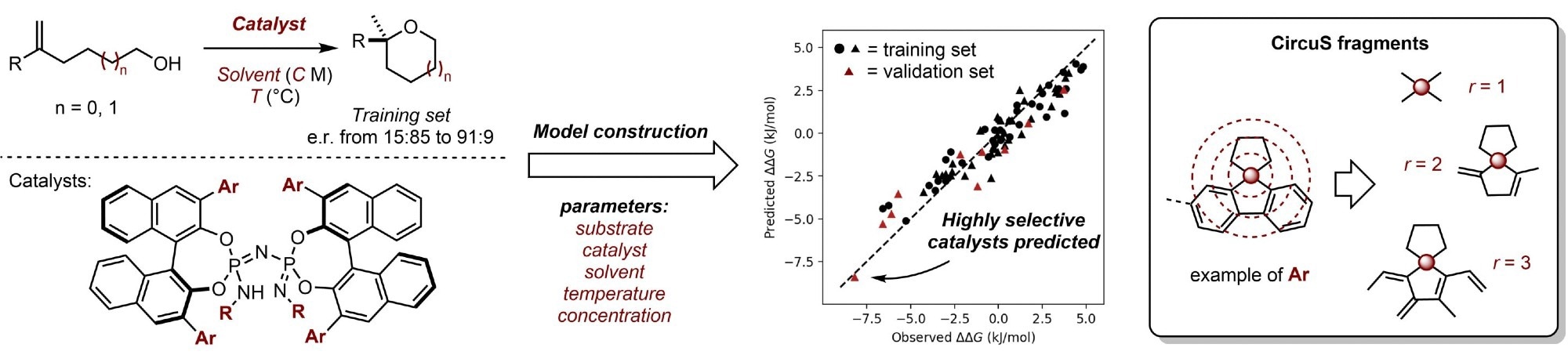
Fast and robust predictive models using 2D descriptors particularly suited for asymmetric catalysis. Highly selective catalysts were predicted and validated using training data with only moderate selectivities. Credit: Nobuya Tsuji, Pavel Sidorov, et al. Angewandte Chemie International Edition. 2023
A study published in the journal Angewandte Chemie International Edition illustrated a machine learning strategy that uses sophisticated but efficient two-dimensional molecular descriptors to reliably predict highly selective asymmetric catalysts without quantum chemical computations.
Traditional Approaches of Predicting Catalysts
Screening techniques for sufficient selectivity in asymmetric catalysis entail significant time and effort and often depend on trial-and-error tests.
Chemists often make educated guesses to determine ideal catalysts with excellent enantioselectivities by taking into account the conformational, electronic, and steric effects of the catalysts.
However, the efficacy of this approach is highly dependent on the chemists involved, and even if these guesses work, the results are usually more qualitative in character. Providing tools that allow for quantitative analysis of screening results is therefore imperative.
Machine Learning-based Models for Catalyst Prediction
Machine learning is regarded as a useful tool for quantifying molecular features like chemical synthesis or biological activity.
Quantitative accounts and estimates of selectivity and reactivity based on structural details use Hammett's method. Here, the electronic and steric characteristics of substituents at certain places in a sequence of catalysts or substrates serve as equation parameters.
The variables are acquired via experiments or quantum chemical computing and are commonly used to optimize catalysis, including asymmetric catalysis, via multivariate linear regression models or non-linear models.
While this technique may give insights into the reaction process, due to restricted computation resources, selecting such descriptors often depends on chemist expertise, making the model incapable of incorporating generic aspects of the catalyst structure.
Pros and Cons of 3D Descriptors
Three-dimensional descriptors provide general structural detail, so models based on 3D structures can find correlations without fully understanding the response mechanism.
The downside of such approaches is that they need expensive quantum chemical computations and, for grid-based systems, core structure alignment.
Numerous 3D structure-based approaches that do not need alignment have been reported in recent literature; however, their effectiveness is limited, particularly in extrapolation.
The Advantages of 2D Descriptors
Since 2D descriptors like binary fingerprints or fragment counts are obtained directly from a two-dimensional representation of a molecule, they also convey general structural properties, although frequently implicitly, and help to avoid expensive computations.
2D descriptors, therefore, offer an obvious speed benefit. Nevertheless, fingerprint descriptors alone are deemed inadequate for capturing the structures required to build reliable prediction models in asymmetric catalysis.
Several fingerprint traits were recently combined to create predictive models. Despite the reasonably strong performance shown by the test set on substrates, the catalyst validation sets still have a scope for improvement, confirming the intrinsic challenge of expressing complex catalytic structures purely using binary 2D fingerprints.
ISIDA (In SIlico design and Data Analysis) platform fragment count descriptors can encode fragments into non-binary vectors depending on the frequency of occurrence in a molecule without reducing the number of features.
ISIDA descriptors provide a wide range of possible chemical structure representations, including fragment size and topology, allowing them to be fine-tuned for the task at hand.
The ISIDA platform allows the computation of fragments for reaction schemes using Condensed Graphs of Reaction (CGR), a unique aspect of this technique that integrates products and reactants into a singular pseudo-molecule having dynamic bonds, which may change as the reaction progresses.
Even though these descriptors have been used to predict pharmacological characteristics, chemical transformations, and materials, there are no reports on their use in asymmetric catalysis.
Highlights of the Study
In this study, the researchers provided a model for predicting the enantioselectivity of structurally diversified and flexible catalysts. The predictive model was based on fragment count descriptors and did not need any quantum chemical computations.
To provide a more exact representation of polyaromatic or cyclic hydrocarbon substituents, which are frequently found in asymmetric catalytic processes, another fragment type was introduced.
The model’s applicability to an actual synthesis problem was also proven in the study. The predictive model was used to identify highly selective catalysts for the asymmetric synthesis of 2,2-disubstituted tetrahydropyran using training data containing only moderately selective catalysts.
Benefits of the Training Model
Pavel Sidorov, a joint first author of the study, commented on the advantages of their training model:
To predict new selective catalysts chemists would use models based on quantum chemical calculations. However, such models are computationally costly, and when the number of compounds and the size of molecules increases, their application becomes limited.
He added, “Models based on 2D structures are much cheaper and therefore can process hundreds and thousands of molecules in seconds. This allows chemists to filter out the compounds they may not be interested in much more quickly.”
Reference
Tsuji, N., Sidorov, P., Zhu, C., Nagata, Y., Gimadiev, T., Varnek, A., & List, B. (2023). Predicting Highly Enantioselective Catalysts Using Tunable Fragment Descriptors. Angewandte Chemie International Edition. Available at: https://doi.org/10.1002/anie.202218659
Disclaimer: The views expressed here are those of the author expressed in their private capacity and do not necessarily represent the views of AZoM.com Limited T/A AZoNetwork the owner and operator of this website. This disclaimer forms part of the Terms and conditions of use of this website.